Polymer Dynamics Stress Production and Relaxation
Project Sponsors
National Science Foundation
Award Number: CMS - 9908025
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science FoundationFaculty Participants
Catalin R. Picu
Professor
Current and Former Students
Mihai C. Pavel
Graduate Research Assistant (2000-2002)
Summary
Some of the basic assumptions made in the molecular theory of polymer dynamics are compared with results obtained from atomistic simulations. In this project we develop a unified description of stress production and relaxation in polymeric systems which attempts to scale-link the atomic and molecular levels, and provides a framework in which the constitutive laws defined at larger scales are based on atomic-scale rather than on molecular-scale processes. This formalism aims at unifying the description of the fast beta-relaxation modes with that of the slower short-time alpha-modes. The fast modes are related to local atomic rearrangements and are not captured by molecular scale descriptions. The slow modes are related to bond/segment re-orientation. The new formalism being developed includes both bonded and non-bonded interactions on an equal footing and is equally valid for short and long chain molecules.
Introduction
Motivation
Polymeric materials are some of the most used materials in today’s technology. They are utilized in the semi-crystalline form, as amorphous glasses and as rubbery elastomers. The diverse and, at times, unusual properties of polymers make them an object of high practical interest subject to continuously increasing research attention.
Despite the remarkable advances made since the inception of the field about 70 years ago, many of the practical problems related to injection and molding still await a sound theoretical treatment that would guide the choices made by process engineers. There are well-known difficulties in designing a reliable molding process due to the complex mechanical behavior of the polymeric melt and solid. Die exit instabilities leading to cracked surfaces, post-extrusion swelling and orientation, and memory effects leading to a high processing history sensitivity are some of the most important issues that have to be solved. Knowledge of the stress history in the melt as well as the melt temperature are important in controlling swelling and orientation at die exit and, ultimately, the stress frozen in the solid material. If the flow separates in passing an obstacle and re-welds downstream, the heterogeneous history in the region of the weld may be remembered and result in an appearance defect, especially in crystalline polymers where melts of different mechanical history may crystallize differently.
Remarkably, there is only one physical phenomenon that underlies swelling, orientation, and memory effects: the relaxation of stresses that build up in the polymeric melt during flow. This demonstrates the importance of rheological studies of these systems.
Stress Relaxation
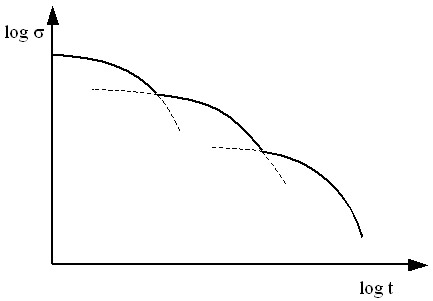
Stress relaxes by a superposition of modes. At high frequencies, in a dynamic test, and at early times, in a relaxation test, beta-relaxation is believed to involve rotation of the side-groups about the chain backbone as well as other fast rearrangements on the atomic scale. Multiple beta modes may exist. At lower frequencies or later times, the relaxation modes are known as alpha-relaxation, and are generally associated with bond and chain segments reorientation. At later times and in highly entangled melts, mass transport through diffusion of whole chains induces additional relaxation.
Figure 1 shows qualitatively the time evolution of stress during relaxation, in a log-log coordinate system. The contribution of the various modes are clearly delineated.
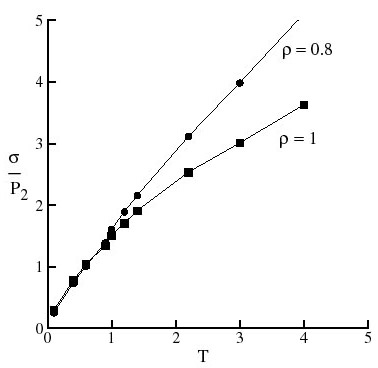
Stress-optical Coefficient
The refraction index of a transparent polymer depends (among other factors) on its internal structure, i.e. on the orientation and distribution of the chains. In the unloaded state, the chains are randomly oriented and the material is isotropic optically. When stretched, chains align and the polymer becomes birefringent. Interestingly, it is observed experimentally that during the alpha-relaxation regime and for small deformations, the birefringence is proportional to the stress. This relation is quantified by the stress-optical coefficient (SOC) or the ratio of birefringence to stress.
Molecular Theory
the Entropic Spring model
Features
In order to describe stress production in polymeric systems, one imagines the following physical picture: at low strains, the environment of a given atom remains essentially unchanged during stretching. This suggests that energetic interactions are not significantly affected by strain as is the case, for instance, in metals. At the same time however, chains stretch to accommodate the imposed deformation. They become hence preferentially oriented in the stretch direction and the entropy of the system decreases. This generates a thermodynamic driving force for the rebound of the chains and hence a stress in the material. This is, in essence, the Entropic Spring model for the chains. It predicts that, at imposed stretch, stress is proportional to the temperature.
This picture incorporates a number of other assumptions. It is for instance assumed that non-bonded interactions do not contribute to stress, which is due to bonded interactions only. Since the overall stress is (say) tensile, this implies that the chains are in tension.
Stress relaxation corresponds in this description to the return to isotropy of chain segment orientation.
Limitations
For a chain (or a segment of a chain) to be statistically regarded as an entropic spring, it must be long enough. The picture has therefore a lower bound length scale. This implies that any structural change which has an intrinsic wavelength smaller than this cut-off cannot be represented and is assumed not to contribute to the overall stress.
Approach
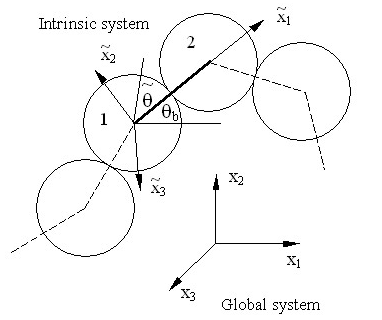
This investigation requires extensive molecular dynamics simulations of model polymeric systems. We have used a united atom model of the “pearl-necklace” type to simulate macromolecules having small side groups (Figure 2). The covalent bonds along the chains are simulated by temperature-insensitive springs of large elastic constant, while the interactions between atoms which are not covalently bonded are assumed to be of the van der Waals type and are simulated by a Lennard-Jones potential.
The simulations are performed using periodic boundary conditions, as customary in molecular dynamics models. Both equilibrium and non-equilibrium conditions are considered. The non-equilibrium state is induced by extensionally deforming the simulation cell while preserving its volume (Figure 3). The deformation is imposed simply by changing the shape of the cell and is transmitted inside it through the periodic boundary conditions imposed on the system. The simulations are performed in the NVT ensemble.
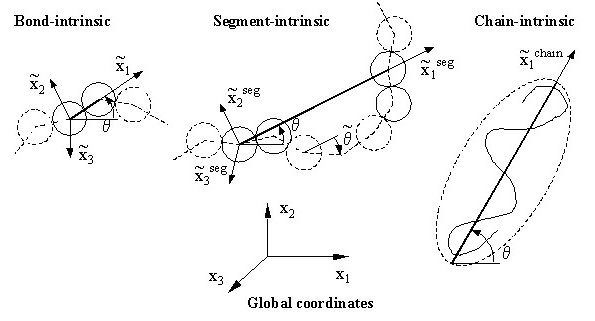
Intrinsic Coordinate System
Relevant statistical parameters are computed from these simulations. Stress is computed using the virial formula in the coordinate system of the simulation cell (global) as well as in an intrinsic coordinate system tied to and which rotates with each bond (Figure 2).
Results
Atom-based Stress
The stress computed at the atomic scale is due to both bonded and non-bonded interactions. The magnitude of these two components is comparable. This contradicts the Entropic Spring model in which it is assumed that non-bonded interactions do not generate deviatoric stress (Figure 4).
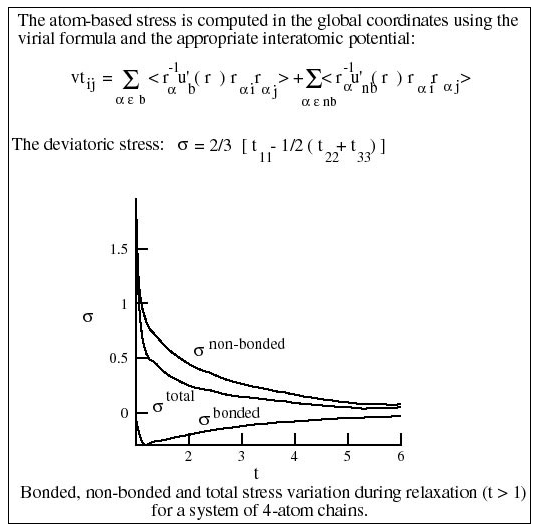
The bonded interactions lead to a negative stress, as opposed to the assumption made in the molecular scale model (Figure 4).
Intrinsic Stress
The intrinsic stress tensor is largely insensitive to melt deformation. Figure 5 shows the variation of this quantity in time during relaxation. Intrinsic stresses are denoted the tilda. During this time, the global stress changes as shown in Figure 4.
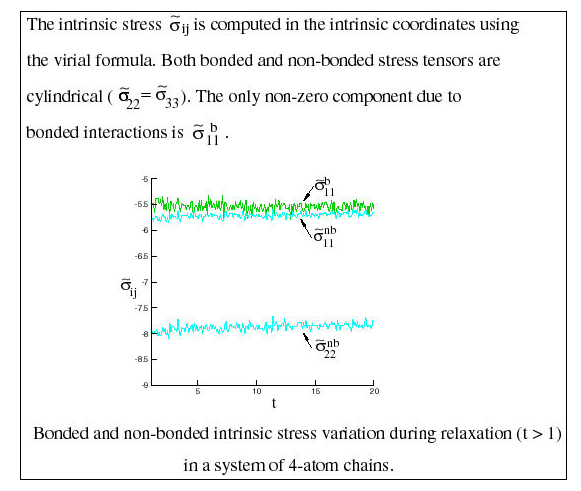
Since the intrinsic stress is an invariant to deformation at small deformations, the stress-optical coefficient may be expressed in terms of the components of this tensor and hence may be computed from an equilibrium simulation.
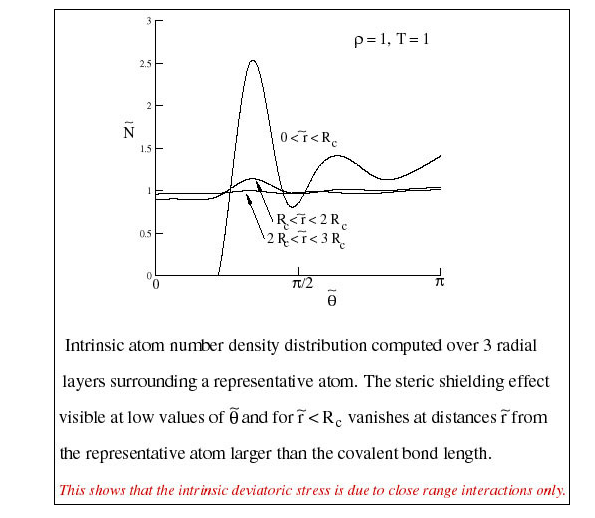
Intrinsic Distribution
The atom number density about of a representative atom may be computed in the intrinsic coordinate system. The distribution is non-uniform due to steric shielding. When computed from non-bonded neighbors that are within a bond length from the representative atom, the distribution looks very similar to the familiar radial distribution function (Figure 6). Note that here the x-axis is the angular position. Interesting, this curve may be used to monitor the glass transition, just as \(g(r)\) is used.
Scale Invariance of the Stress Production Mechanism in Polymeric Systems
A physical description of stress production that unifies the bond, segment and the chain scales within the Intrinsic Stress Framework has been proposed. The stress is produced by bonded and non-bonded interatomic interactions on the scale of a bead. The global, system level stress results by averaging the atomic level stress in global coordinates. The atomic level stress may be also computed in a family of mobile frames tied to the generic bond, to the generic chain segment, or to the representative ellipsoidal coil (Fig. 7). These intrinsic stresses have significant deviatoric components. We have shown that the bond, the chain segment and the whole chain carry the same deviatoric stress (Figs. 8 and 9). The hydrostatic component of the intrinsic stress scales with the segment length.
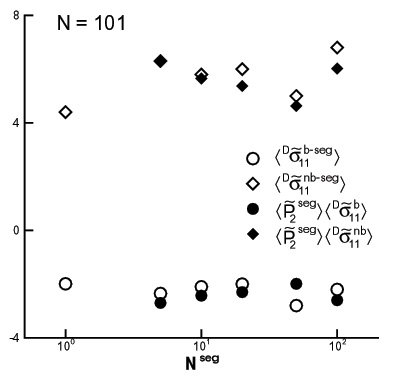
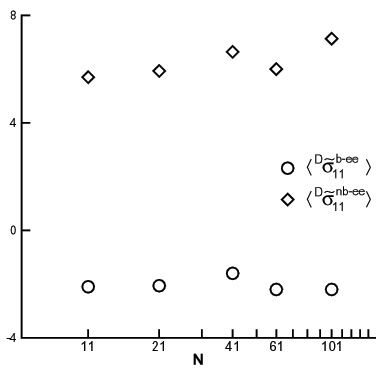
The scale invariance of the deviatoric intrinsic stress allows for the development of a unitary description of stress production. In the Intrinsic Stress Framework, the global deviatoric stress is seen as being generated by the preferential orientation of bond-intrinsic frames each carrying a non-zero deviatoric intrinsic stress which is independent of deformation. This physical picture is translated here to larger scales by showing that the global deviatoric stress can be similarly viewed as being associated with the preferential orientation of larger units (segments or chains), which carry a non-zero deviatoric stress. The extension is valid provided the intrinsic stress computed on larger scales (segments or chains) is deformation insensitive, as is the case with the bond-scale intrinsic stress tensor. We have shown that this situation exists late in the relaxation history.
The reorientation of segments (and chains) during a relaxation is described by processes that take place with different rates. During the initial stage of relaxation the internal chain structure (characterized by chain-intrinsic parameters) returns to its equilibrium configuration. This process corresponds to the fast Rouse modes. Once the size of the ellipsoidal coil and the orientation of bonds with respect to the coil axis have equilibrated, the chains rotate to randomize the chain orientation in the global coordinate system. The collective rotation of the chains is described by an exponential decay. Once the first regime completes, the chain scale intrinsic stress reaches the equilibrium value. The physical picture of global stress production described here applies for the second regime.
The Entropic Nature of Stress
The molecular scale stress is entropic by definition. It has been proved by means of simulations that the three non-zero components of the intrinsic stress tensor are not purely entropic. However, their energetic components cancel each other such that the global stress is purely entropic (Figure 10). It is interesting that the source of this entropic behavior is the packing entropy (just like in simple liquids) rather than the configurational entropy of the chains as a whole.
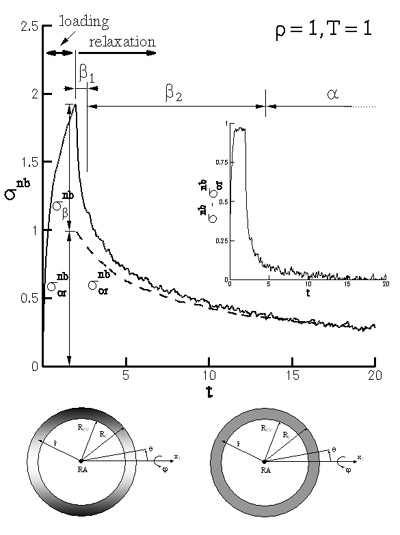
Fast Stress Relaxation in Polymer Melts
The fast \(\beta\) stress relaxation modes in a model polymeric melt are investigated by means of non-equilibrium molecular dynamics simulations. The stress is computed on the atomic level by accounting for both bonded and non-bonded interatomic interactions. Stress relaxation takes place by several modes (Figure 11), each corresponding to specific atomic scale structural changes. The \(\beta\) relaxation corresponds to the return to isotropy of the atomic distribution in the neighborhood of a representative atom RA, and encompasses a quasi-elastic mode (\(\beta_1\)), and a slower mode (\(\beta_2\)). The neighbor distribution about the representative atom (after subtracting the contribution of preferential bond orientation - “corrected distribution”) is shown schematically in Figure 11. At the beginning of relaxation the distribution has a cylindrical symmetry about the stretch direction \(x_1\). At \(t = 14\) the corrected distribution is isotropic and the global stress is due to preferential bond orientation only.
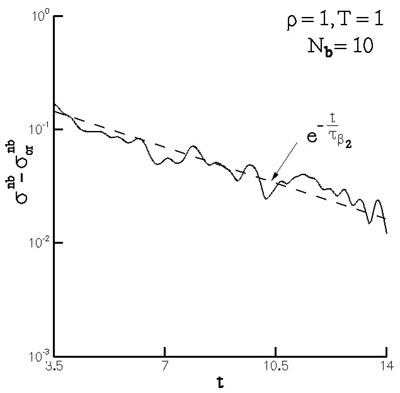
The \(\beta_2\) mode corresponds to the isotropization of the neighbor density about the representative atom as shown in Figure 11, and may be described by a diffusion-like process. The mode is exponential. Figure 12 shows the corrected stress difference during this regime, while Figure 13 shows the relaxation of a measure of the neighbor number density. The two relaxation processes may be described by the same time constant which substantiates the connection between the diffusion-like process and stress relaxation.
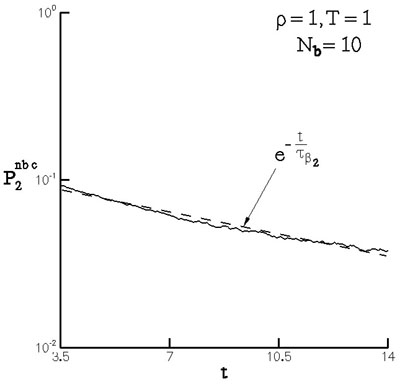
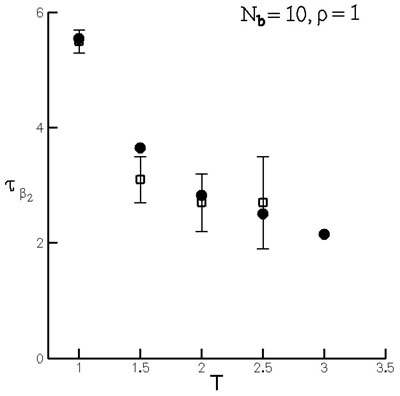
The time constant for the exponential mode (\(\beta_2\)) may be derived from equilibrium simulations (Figure 14) based on the fluctuation dissipation theorem and a continuum model of diffusion in the neighborhood of a representative atom. This shows that the non-equilibrium system is in the linear response regime during this relaxation period.
References
- [1] introduced the concept of intrinsic atomic-level stress.
- [2], [3] discuss the validity of the Entropic Spring model and review earlier work.
- [4] develops concepts related to those discussed here.
- [1]J. Gao and J. H. Weiner, “Intrinsic atomic‐level stresses in polymer melts and networks,” The Journal of Chemical Physics, vol. 90, no. 11, pp. 6749–6760, Jun. 1989, doi: 10.1063/1.456682.
By means of the virial stress formula, the macroscopic stress tensor in a polymer system may be expressed as a sum of atomic‐level stress tensors, with each of the latter associated with a single atom and with all tensors, macroscopic and atomic, referred to the same fixed laboratory reference frame. In order to gain insight into the interplay between the covalent and noncovalent interactions in such systems, we refer the atomic‐level stress tensor associated with a given atom to a moving local frame which maintains a fixed orientation with respect to the covalent structure attached to that atom; we term this the intrinsic atomic‐level stress. We compute, by the method of molecular dynamics, the intrinsic stresses for model polymer melts and networks based on systems of freely rotating chains. The noncovalent interactions are through a truncated Lennard‐Jones potential which is taken as either purely repulsive or has an attractive portion. We observe that (i) the intrinsic atomic‐level stresses are highly anisotropic, even in an equilibrium melt in which the macroscopic stress is isotropic; (ii) the intrinsic atomic‐level stress is the same in a polymer melt and in the corresponding polymer network model, if the latter is only moderately deformed; (iii) the intrinsic stresses in dense polymer melts are very different in the presence of noncovalent interactions from those in ideal systems; (iv) the addition of an attractive portion to the noncovalent potential has large effects on the deviatoric part of the intrinsic stress, thus casting doubt on the utility of the van der Waals’ picture for such systems.
- [2]J. Gao and J. H. Weiner, “Nature of Stress on the Atomic Level in Dense Polymer Systems,” Science, vol. 266, no. 5186, pp. 748–752, Nov. 1994, doi: 10.1126/science.266.5186.748.
Stress in dense polymer systems is classically viewed as being molecular in character and is based on the entropic spring concept. A description on the atomic level has been developed on the basis of extensive computer simulations. An important new concept is the intrinsic monomer stress (IMS), the individual monomer contribution to the macroscopic stress referred to a local moving coordinate system in which the backbone bonds attached to that monomer are fixed. The IMS is time-independent and, for a given polymer system at fixed density, has the same value in the equilibrium melt, with the melt undergoing stress relaxation, and in the deformed cross-linked system.
- [3]J. Gao and J. H. Weiner, “Range of validity of the entropic spring concept in polymer melt relaxation,” Macromolecules, vol. 25, no. 13, pp. 3462–3467, Jun. 1992, doi: 10.1021/ma00039a024.
- [4]M. Kröger, C. Luap, and R. Muller, “Polymer Melts under Uniaxial Elongational Flow: Stress−Optical Behavior from Experiments and Nonequilibrium Molecular Dynamics Computer Simulations,” Macromolecules, vol. 30, no. 3, pp. 526–539, Feb. 1997, doi: 10.1021/ma960317c.
Tensile stress and birefringence in both real and model amorphous polymer melts have been measured during constant rate uniaxial elongational flow. We focus on investigations where deviations from the linear stress?optical behavior are pronounced. A rate-dependent contribution to the stress which is not directly related to the intramolecular conformations (?stress offset?) is detected for both types of macromolecular fluids. Independent of the flow history, during relaxation a linear stress?optical behavior is revealed. Nonequilibrium molecular dynamics (NEMD) computer simulations on the multibead anharmonic spring model are shown to provide insight into the molecular mechanisms underlying the viscoelastic behavior:? during relaxation the intermolecular interactions become dominant in correlation with linear stress?optical behavior; the stress offset is shown to be very similar to the stress arising in the corresponding simple fluid; the total stress can well be approximated by a sum of three parts which are based on single-particle and single-link distribution functions only; the yield point behavior at high elongation rates reflects the transition from affine to nonaffine motion of bonds and is understood without reference to strong inhomogeneities resulting from local plastic strain production [the chemical structure does not influence the qualitative behavior]; distinct microscopic stress contributions under elongation and subsequent relaxation such as inter- and intramolecular, attractive and repulsive, kinetic and potential contributions are resolved.
Publications
- [1]C. R. Picu and M. C. Pavel, “Scale invariance of the stress production mechanism in polymeric systems,” Macromolecules, vol. 36, no. 24, pp. 9205–9215, 2003, Accessed: Jan. 24, 2016. [Online]. Available at: http://pubs.acs.org/doi/abs/10.1021/ma0259867.
Stress production in a model monodisperse polymeric material is investigated on multiple scales. The analysis is performed by means of equilibrium and non-equilibrium molecular dynamics. A family of mobile intrinsic coordinate systems is introduced, each system having one axis tied to the end-to-end vector of a generic chain segment of specified length. A similar mobile coordinate system tied to the large semiaxis of the ellipsoidal chains is defined on the chain scale. The atomic level stress is evaluated based on bonded and non-bondedinteratomic interactions, and averaged in the global coordinate system, to result in the global, system level stress, and in the various intrinsic systems, to result in intrinsic stresses. It is observed that the deviatoric intrinsic stress is scale independent, a bond, a chain segment and the chain scale intrinsic frame carrying the same stress. The hydrostatic component of the stress tensor scales with the segment length. This concept extends the previously introduced Intrinsic Stress Framework, scale linking the bond and chain scales. During melt deformation, the chain segments stretch and rotate. Chains shorter than the entanglement length mainly rotate during an elongational deformation of limited amplitude, their size remaining essentially constant. Longer chains distort and rotate. Two regimes are evidenced during the return to isotropy of the orientation on multiple scales. The faster mode is associated with the return to equilibrium of the internal structure of the generic chain, while the slower mode is associated with chain rotation in the global coordinate system. The intrinsic deviatoric stress carried by a chain changes during the first mode, and is essentially constant during the second. The physical picture of stress production defined on the scale of a bond (Kuhn segment) in the Intrinsic Stress Framework translates to the chain scale during this late relaxation regime: each rotating chain carries a constant deviatoric intrinsic stress, the preferential chain orientation leading to a non-zero global deviatoric stress.
- [2]C. R. Picu and M. C. Pavel, “Fast relaxation modes in model polymeric systems,” Macromolecules, vol. 35, no. 5, pp. 1840–1847, 2002, Accessed: Jan. 24, 2016. [Online]. Available at: http://pubs.acs.org/doi/abs/10.1021/ma0115949.
- [3]C. R. Picu, “Entropic character of the atomic level stress in polymeric melts,” Macromolecules, vol. 34, no. 14, pp. 5023–5029, 2001, Accessed: Jan. 24, 2016. [Online]. Available at: http://pubs.acs.org/doi/abs/10.1021/ma002186s.
- [4]C. R. Picu, G. Loriot, and J. H. Weiner, “Toward a unified view of stress in small-molecular and in macromolecular liquids,” The Journal of chemical physics, vol. 110, no. 9, pp. 4678–4686, 1999, Accessed: Jan. 24, 2016. [Online]. Available at: http://scitation.aip.org/content/aip/journal/jcp/110/9/10.1063/1.478351.
- [5]C. R. Picu, “Intrinsic distribution and atomic level stress in polymeric melts,” Macromolecules, vol. 32, no. 21, pp. 7319–7324, 1999, Accessed: Jan. 24, 2016. [Online]. Available at: http://pubs.acs.org/doi/abs/10.1021/ma990836q.
- [6]C. R. Picu and J. H. Weiner, “Stress relaxation in a diatomic liquid,” The Journal of Chemical Physics, vol. 108, no. 12, pp. 4984–4991, Mar. 1998, doi: 10.1063/1.475907.
- [7]C. R. Picu and J. H. Weiner, “Structural changes during stress relaxation in simple liquids,” The Journal of chemical physics, vol. 107, no. 18, pp. 7214–7222, 1997, Accessed: Jan. 24, 2016. [Online]. Available at: http://scitation.aip.org/content/aip/journal/jcp/107/18/10.1063/1.474962.